概述
本项目旨在通过人工智能与合成生物学的交叉学科研究,建立枯草芽孢杆菌从头合成启动子的设计与启动子强度分析系统,并实现生成特定强度的人工启动子。
经过前期的调研和讨论,我们分别使用两种方式生成启动子:通过缩小重构误差、提高分布相似性来学习训练样本特征的变分自编码器(Variational auto-encoder,VAE),和基于生物界中的“自然选择”思想的遗传算法(Genetic Algorithm,GA)。另一方面,我们还构建了一个强度预测网络,通过将其与生成模型组合使用,对生成的启动子进行约束,从而获得目标强度的启动子。整体模型框架如下图1所示。
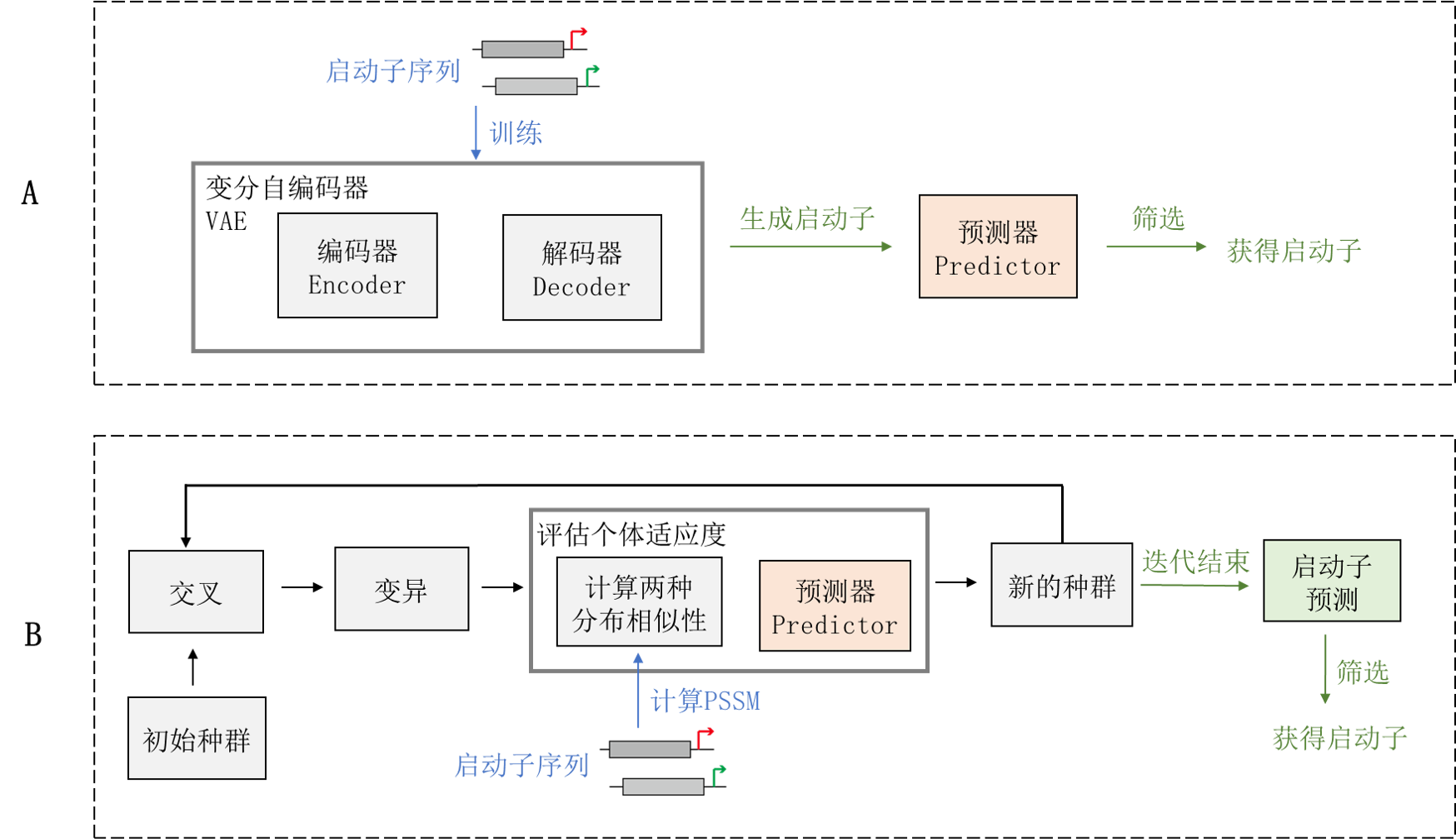
图1 两种方式生成特定强度的启动子
A:基于神经网络的模型框架,首先由VAE生成大量启动子,再使用预测器进行强度筛选;B:基于遗传算法的模型框架,通过将预测器预测结果作为目标函数之一,在每轮迭代过程中不断进行强度的筛选,最后将生成的启动子利用启动子预测软件进行过滤,以提高生成结果的质量。
启动子强度预测模型
利用启动子序列信息实现启动子强度的定量预测需要挖掘出序列中决定启动子强度的“特征”信息。本项目利用深度学习技术构建从序列预测启动子强度的回归模型,利用卷积神经网络进行对序列特征进行提取与学习,利用长短期记忆神经网络对功能域之间的长期依赖关系进行学习。模型主要包括嵌入阶段(Embeding stage)、卷积阶段(CNN stage)、长短期记忆神经网络阶段(LSTM stage)、密集化阶段(Dense stage)等结构,接收输入的启动子序列,分别进行数据转换和特征提取,并最终输出启动子的强度预测值。
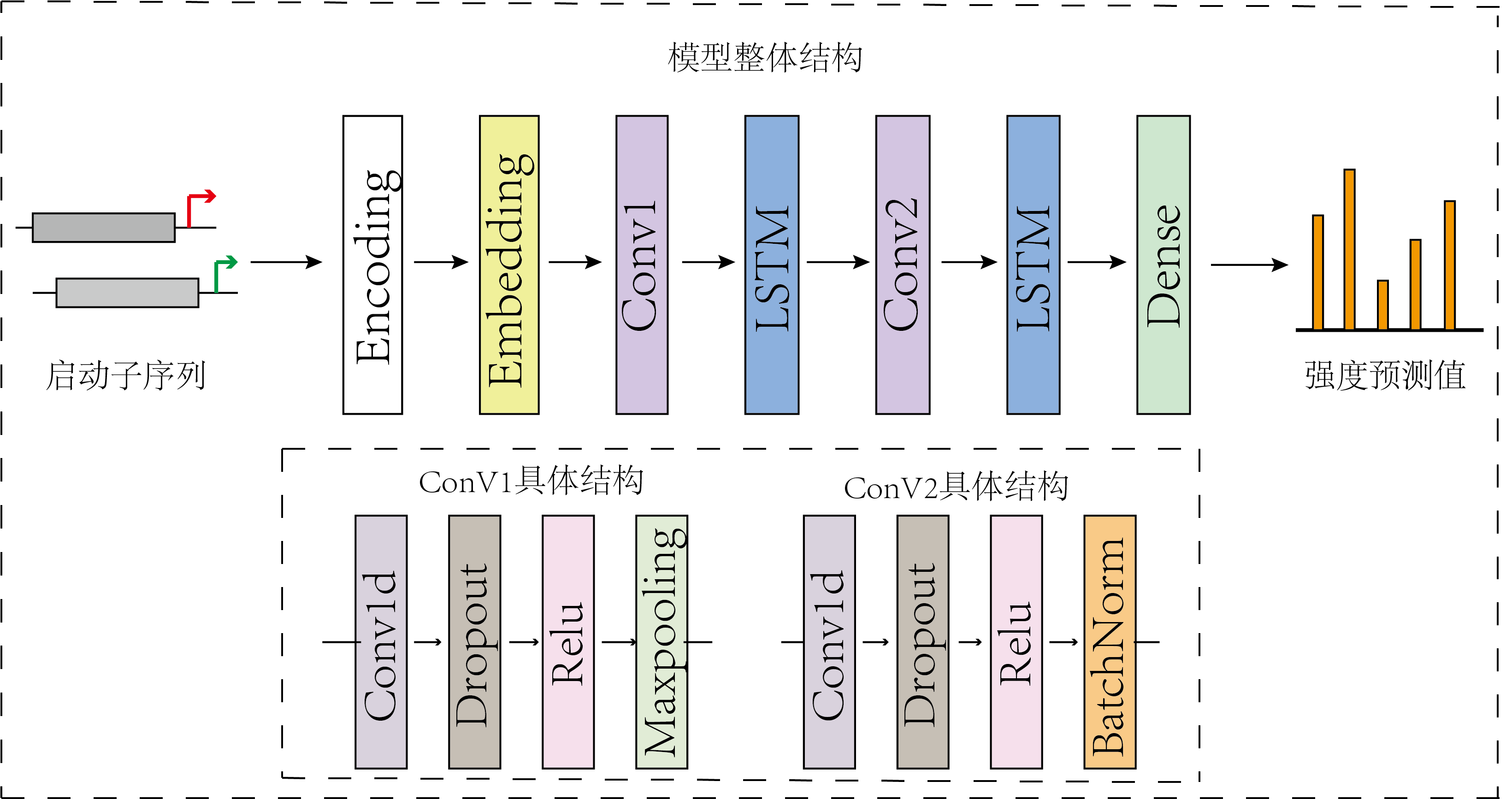
图2 强度预测模型结构图
(1)数据预处理
本项目针对枯草芽孢杆菌启动子,设定的启动子序列长度为61bp。为了使启动子序列能被计算机识别,工作的第一步就是对序列中的A、T、C、G的碱基信息进行编码(encoding),按{A: 1, T: 2, C: 3, G: 4}的规则,将四个碱基序列分别映射成一个具体的实数。(编码的顺序单纯地为了方便起见,不影响实验的结果)
(2)嵌入阶段(Embedding stage)
嵌入阶段会将预处理得到的序列向量中的每一个数字映射成一个固定长度(embedding_dim)的向量。假设embedding_dim=16,则一个长度为61的启动子序列在经过预处理和嵌入阶段后,会转化为一个61×16的矩阵,用于后续的特征提取。相比与常用的one-hot编码方式,嵌入阶段的引入能使模型具有更灵活的特征学习与参数调整方案。最终模型选取的embedding_dim=128。
(3)卷积阶段(CNN stage)
卷积阶段会利用多个卷积核去探测上述获得的矩阵,进行特征提取。可以将卷积核看作是motif探测器,利用卷积核探测启动子序列中关键及潜在的基序特征。在卷积操作后,依次引入激活函数Relu以增加模型非线性属性,引入池化层进行下采样,用于特征降维,压缩数据和参数的数量,减小过拟合,同时提高模型的容错性。
(4)长短期记忆神经网络阶段(LSTM stage)
LSTM是循环神经网络RNN的一种性能更好的模型,它将由卷积阶段产生的特征映像以时间步长的形式输入到LSTM神经网络中,输出一个固定长度的向量。其主要用1个特定的记忆细胞去存储信息,对于存在依赖关系的数据有着非常显著的性能。模型通过引入LSTM以加强模型对于基序间依赖关系的发掘,有助于学习到更多序列的潜在特征。
(5)密集化阶段(Dense stage)
在密集化阶段,通过全连接层将上述步骤得到的特征映像成为一个具体的数字,从而作为启动子强度的预测值输出。
>> 具体训练结果 详见 结果页面
基于VAE的启动子生成模型
变分自编码器(Variational auto-encoder,VAE)是一类生成模型,由编码器(encoder)和解码器(decoder)两部分组成,原始数据经过编码器的处理后生成一个隐空间中的隐含向量,而解码器可以识别出这个隐含向量中的信息,将其还原为原始数据。通过直接从特定的分布中采样并使用解码器进行还原,就可以实现与训练样本相似但不同的新数据的生成。VAE的优势在于其可以形成一个连续的隐空间,产生平滑过渡的样本。
对于输入数据,我们选择对61bp的启动子序列使用one-hot的编码方式,并将其转换为图像表示,如图3所示。

图3 启动子序列的图像示意图
每4列分别代表一条序列中的A、C、G、T
我们发现这种图像表示的序列其实已经是一种相对简明的数据编码方式,因此在编码时我们没有再使用卷积层对其进行降维处理,而是直接使用全连接层将这种编码方式映射到隐空间,模型结构如图4示。通过这种方式构建的模型结构简单、训练速度快,但仍拥有较好的性能。
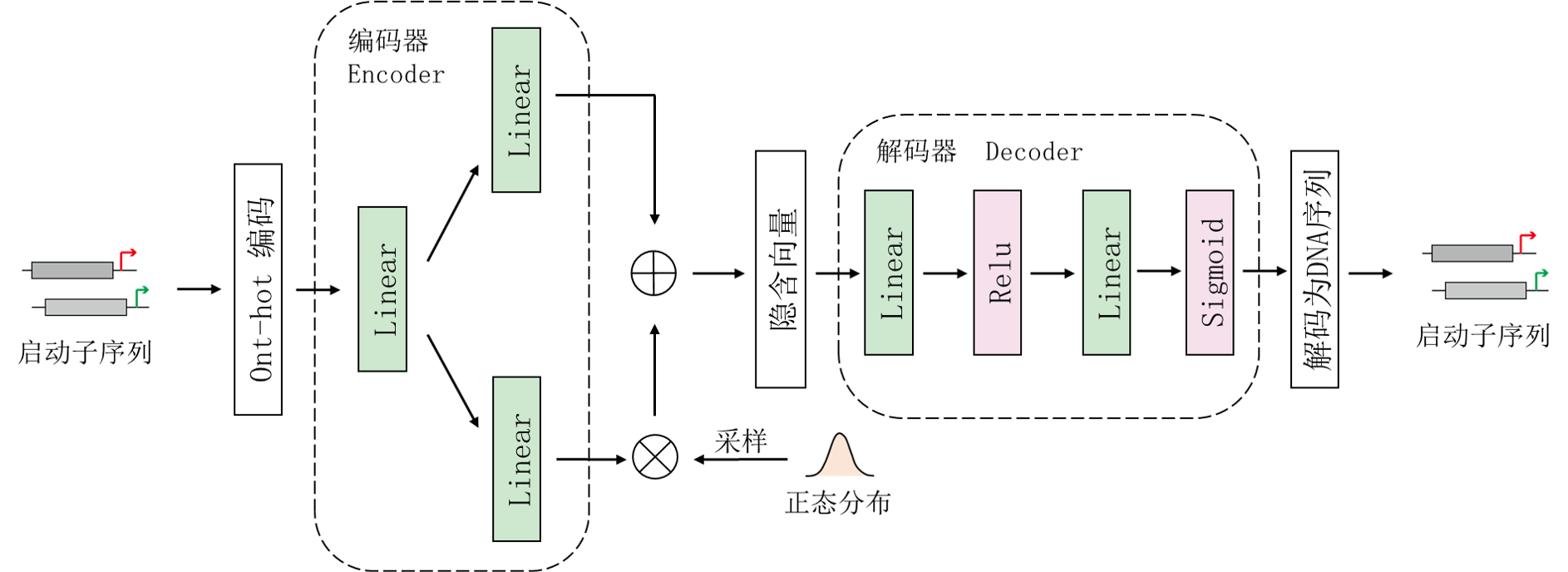
图4 VAE结构示意图
>> 具体训练结果 详见 结果页面
基于遗传算法的启动子生成模型
遗传算法(Genetic Algorithm,GA)是模拟达尔文生物进化论的自然选择和遗传学机理的生物进化过程的计算模型,是一种通过模拟自然进化过程搜索最优解的方法。遗传算法的基本运算过程如下:
首先随机生成序列进行种群初始化。选择个体适应度较高的序列,进行交叉,产生子代,并淘汰达不到强度要求的序列。最后对子代的序列进行变异以确保多样性。重复多次,直到产生达到强度的序列。
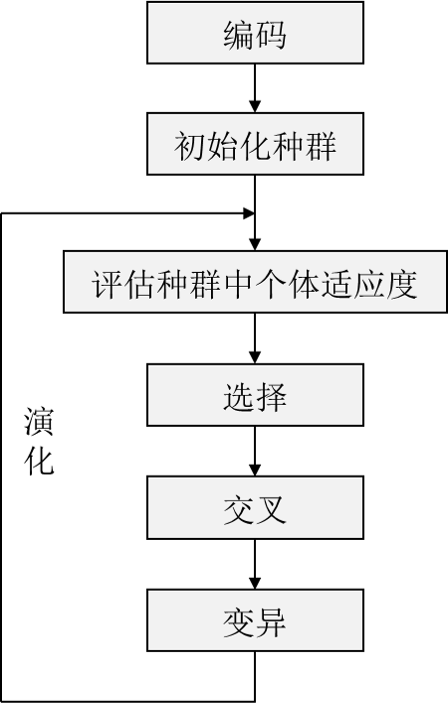
图5 遗传算法的基础思路
在模型的建立过程中,随着评价指标的不断完善,实现了下表中的三个版本:
表1 不同版本的遗传算法
| 版本 | 简介 |
|---|---|
| v1.0 | 评价个体适应度时,仅考虑生成序列与目标序列整体的相似度。 |
| v2.0 | 在v1.0的基础上,考虑生成序列与目标种群的个体差异。 |
| v3.0 | 在v2.0的基础上,对于每一代新种群的每一个个体都预测强度,淘汰强度超出目标范围的个体。 |
遗传算法v1.0中适应函数fitness的计算方式主要围绕位置特定评分矩阵(PSSM)。位置特定评分矩阵(PSSM),也称为位置特定权重矩阵(PSWM)或位置权重矩阵(PWM),是生物序列中基序(模式)motifs(patterns)的常用表示。PSSM的计算方式为:根据多条序列碱基分布计算概率矩阵(PPM),再从概率矩阵(PPM)计算位置特定评分矩阵(PSSM)。
遗传算法v2.0中通过cd-hit-est的方式对不同类别的个体聚类, 计算个体数较多的类的PSSM,每次产生后代时将后代的PSSM与每个类的PSSM比较,淘汰差异过近的后代,这样保证让后代与目标种群的总体PSSM分布差异较小的同时,也保证了不同类别个体的差异度。具体试验过程见下表。
表2 聚类实验
| 试验编号 | 聚类参数 | 参数说明 | 聚类结果 | 评价 |
|---|---|---|---|---|
| 1 | Parameter set -d 0 -n 10 -l 11 -r 1 -p 1 -G 0 -c 0.95 -aS 0.8 | 默认参数 | 4401个clusters | 聚类个数过多,存在许多一个序列自成一个类的情况. |
| 2 | sequence identity threshold = 0.90 | 与试验1相比减小了差异阈值 | 4398个clusters | 聚类个数过多,存在许多一个序列自成一个类的情况 |
| 3 | Parameter set -d 0 -n 10 -l 11 -r 1 -p 1 -G 0 -c 0.95 -aS 0.1 | 与试验1相比aS从0.8改成0.1,增加了比对的局部性 | 2159个clusters | 聚类个数仍然较多 |
| 4 | Parameter set -d 0 -n 10 -l 11 -r 1 -p 1 -G 0 -c 0.8 -aS 0.1 | 与试验1相比c改成了0.8,aS改成了0.1 | 745个clusters | 聚类个数减少,有改善,可以使用,选取前30个最大的类来使用(每个类) |
遗传算法v3.0中考虑了生成序列的强度,这一评价指标通过深度神经网络预测模型产生,最终的算法执行流程如图6所示。
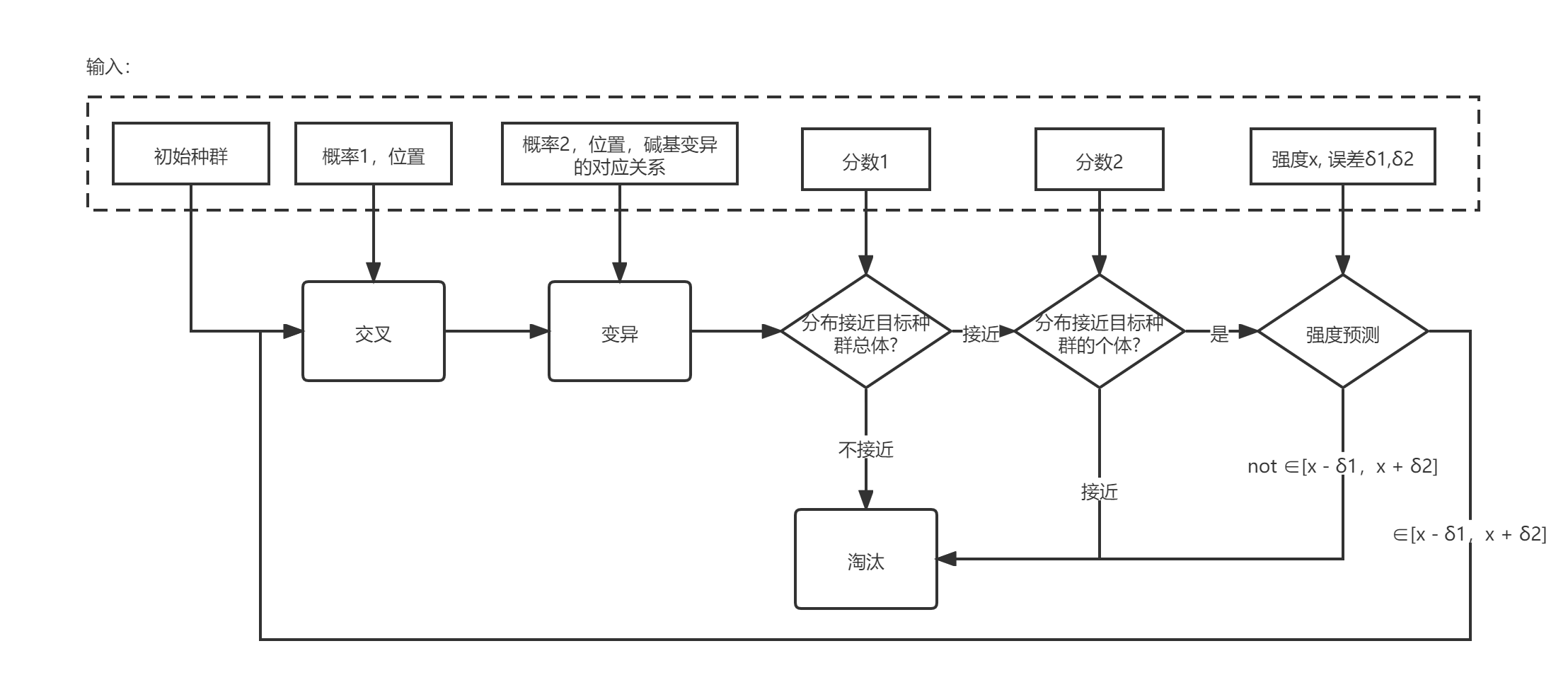
图6 遗传算法执行流程
>> 具体训练结果 详见 结果页面