转录组测序
我们对B. subtilis 168进行了转录组测序,以得到枯草芽孢杆菌中启动子与其转录强度的对应关系,从而构建序列-转录强度数据库,具体实验流程如下:
一、菌体培养及制样
- 挑取平板活化的单克隆菌株接入5 mL LB培养基中,37 ℃、220 r/min条件下过夜培养。
- 按照1%的接种量转入50 mL发酵培养基(20g/L葡萄糖,20g/L胰蛋白胨,0.154g/L NaH2PO4,0.4g/L Na2HPO4·2H2O,0.5g/L MgSO4·7H2O PH=7.2)中,在同样的条件下培养。每组3个平行,待生长至对数生长中期停止生长。
- 取出菌体4 ℃离心快速收集,倒掉上清液,用1mL无菌水重悬菌体并再次离心。
- 液氮速冻并保存于–80 ℃,将样品送至公司进行转录组测序分析。
二、转录组测序分析
转录组测序分析包括RNA提取、RNA样品质量检测、文库构建、文库纯化、文库检测、文库定量、测序簇的生成以及上机测序。
- 提取总RNA。通过RNeasy Mini Kit(Qiagen, Valencia, CA, USA)试剂盒完成。
- RNA质量检测。通过安捷伦2100生物分析仪(Agilent 2100 Bioanalyzer, Agilent)、微量紫外分光光度计(Thermo Fisher Scientific Inc)和1%琼脂糖凝胶对每个样品的总RNA进行定量和鉴定。
- rRNA去除。通过rRNA removal Kit试剂盒进行核糖体RNA的去除
- cDNA的合成。以片段化的mRNA为模板,利用反转录酶SuperScript II和随机引物合成第一链cDNA。第二链cDNA的合成利用第二链合成酶混合物(包括dACG-TP/dUTP)完成。
- cDNA 的末端修饰。利用同时具有3’-5’外切酶和聚合酶活性的End Repair Mix,分别剪切和填补cDNA的3’和5’,使其双端变为平末端。并通过AMPure Xpbeads纯化产物。除此之外,末端修饰还包括3’端腺苷酸化和接头连接。
- PCR扩增富集。将上述获得cDNA进行 PCR 扩增富集。通过Agilent 2100 Bioanalyzer对纯化后的PCR产物进行检测,合格后作为RNA-Seq测序的cDNA库。
- cDNA文库测序分析。依据Illumina Hiseq平台,采用2x150 “pair-end”策略进行文库测序。这个测序由GENEWIZ(中国)公司完成。
- 原始序列数据
- 测序数据质量评估
- 参考序列比对分析
- 新转录本预测
- SNV和InDel分析
- RNA-seq整体量评估
- 基因表达分析
- 基因结构分析
- UTR注释
- sRNA分析
基因组尺度启动子识别及序列-转录强度数据库的建立
由于深度神经网络的训练需要庞大的数据集,实验和文献的数据量不足以支持神经网络的训练。为解决这一问题,首先,在NCBI数据库 (https://www.ncbi.nlm.nih.gov/genome/665?genome_assembly_id=300274) 中找到野生型枯草芽孢杆菌Bacullus subtilis 168中的所有基因,共4536个,并通过爬虫获取其所有基因的上游200bp基因序列。然后通过预测的方法产生基因上游200bp中最有可能是启动子的序列。选取表1报道的软件分别进行预测分析。
| 预测工具 | 网址 |
| iProEP | http://lin-group.cn/server/iProEP/pages/predictor.php |
| Bacpp | http://bacpp.bioinfoucs.com/ferramenta |
| Sph | https://sapphire.biw.kuleuven.be/index.php |
| BDPG | https://www.fruitfly.org/seq_tools/promoter.html |
| Ipro70 | http://ipro70.pythonanywhere.com/server |
其中,对于iProEP,可以选择物种,在这里选择枯草芽孢杆菌。Bacpp中有sigma24,sigma28等多个因子作为选项,Sph中有run SAPPHIRE_1,SAPPHIRE_2_pseudomonas,SAPPHIRE_2_salmonella三个选项。
选择60个已知启动子序列使用上述工具预测,统计多款工具对这些序列的预测结果,计算预测结果与真实启动子位置差值的最大值、最小值、方差和平均值到如下可视化结果。
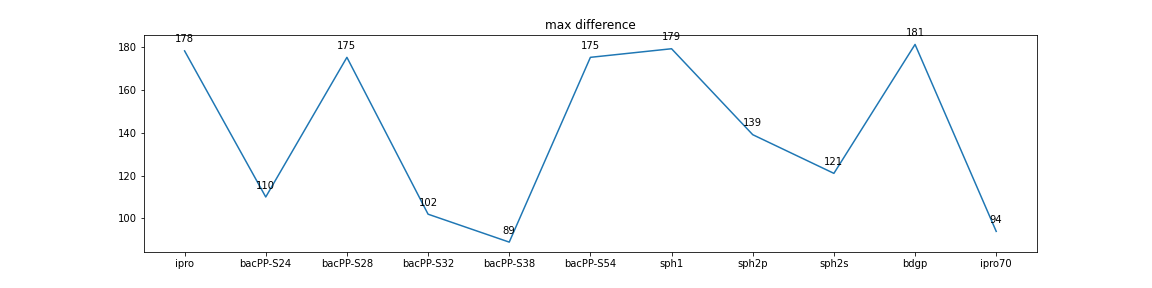

图1 最大值与最小值
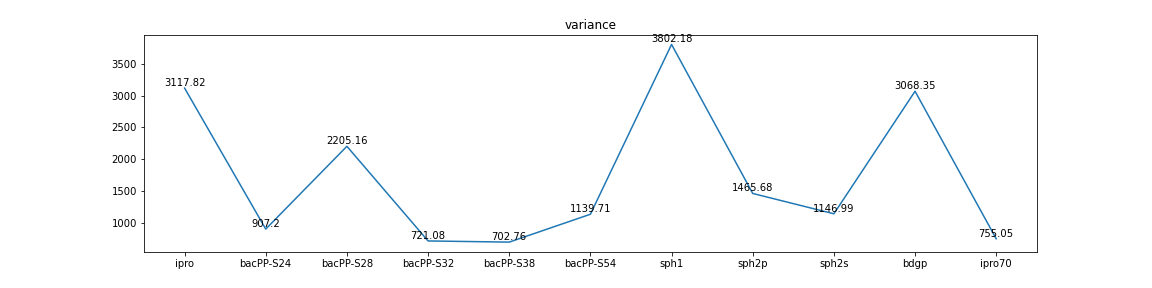
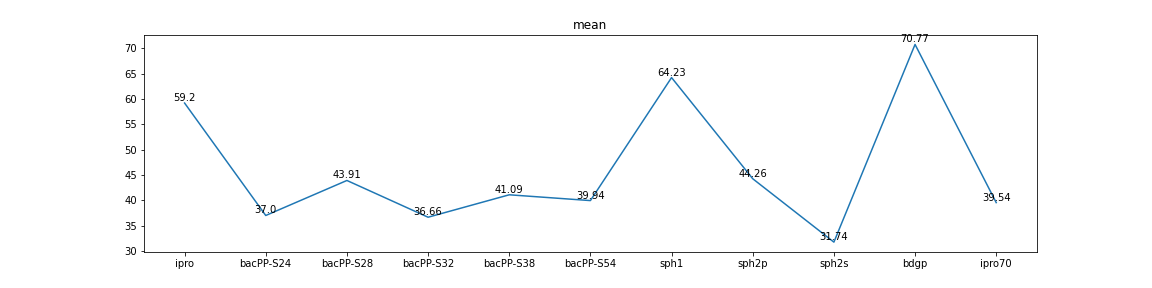
图2 方差和平均值
从以上两图中,我们不难得到如下结果:
- 最大误差(越小越好) sph2s < ipro70 < bacppS38 < bacppS24 < else
- 最小误差(越小越好) sph2s = sph2p = ipro70 = sph1 = else
- 方差(越小越好) sph2s < sph2p < ipro70 < sph1 < else
- 平均值(越小越好) sph2s < sph2p < ipro70 < sph1 < else
- 误差总和(越小越好) sph2s < sph2p < ipro70 < sph1 < else
最终比对多个软件的预测结果,其中 SAPPHIRE 2 for salmonela (沙门氏菌数据训练的人工神经网络)、SAPPHIRE 2 for Pseudomonas (假单胞菌数据训练的人工神经网络)、ipro70 (使用多窗口和最小特征识别Sigma70启动子)的预测效果较好。
在此基础上用这三个工具预测的启动子位置投票出最终结果,投票流程如下:
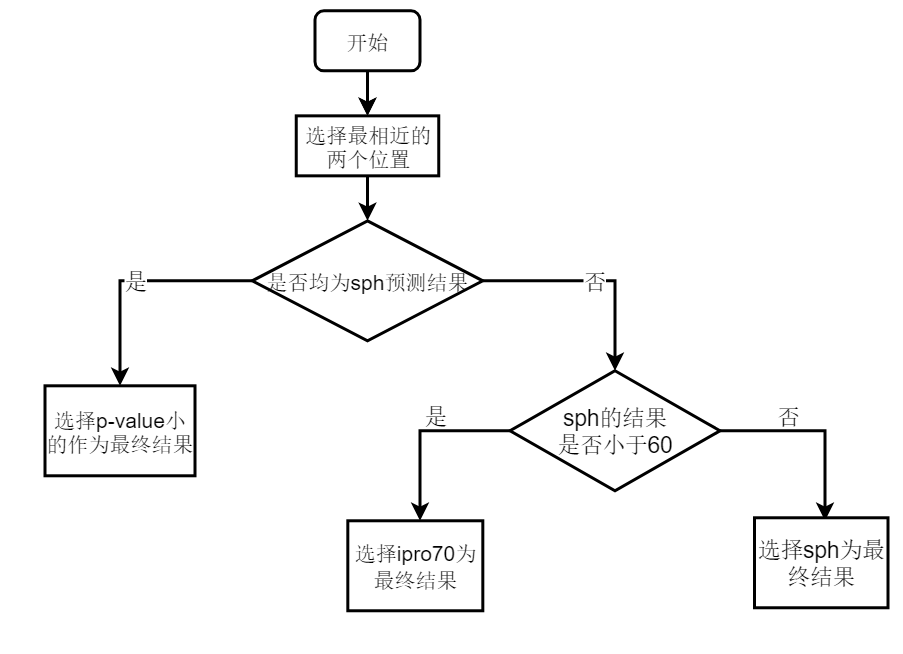
图3 启动子位置投票流程
潜在问题:
- 一个基因对应不只一个启动子(串联启动子的情况,中间会隔几十bp不会有重叠)。
- 一个基因前面可能没有启动子(操纵子的情况)。
应对方式:
- 忽略这种情况,只取一个打分最高的启动子。
- 如果所有预测都打分较低则放弃这个基因。
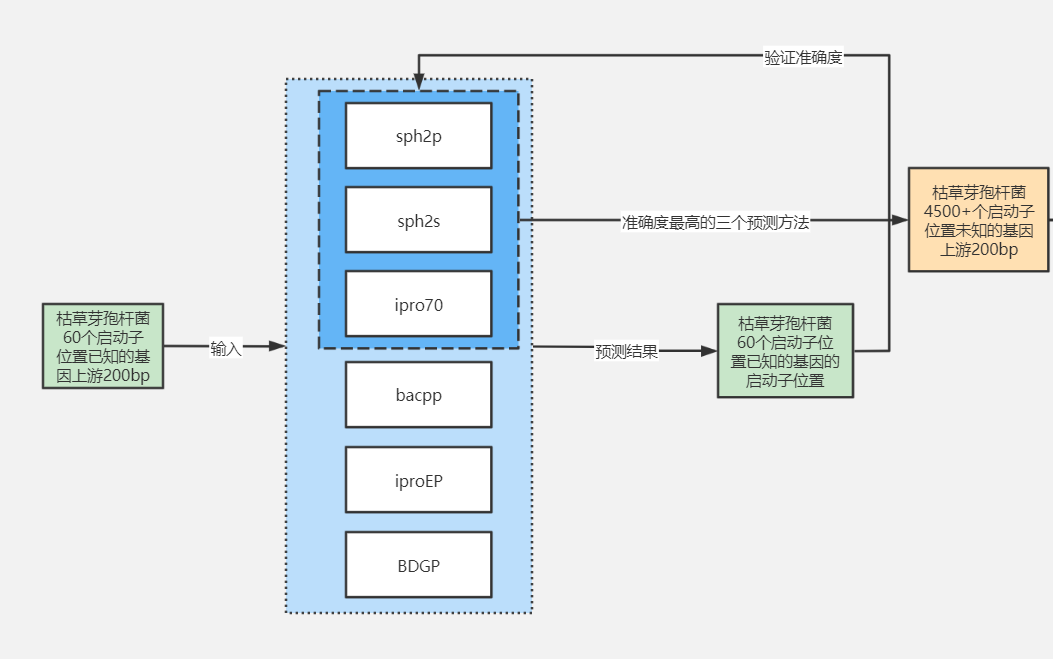
图4 启动子预测数据整合工作流
通过以上步骤,最终得到约4500个枯草芽孢杆菌的预测启动子序列,结合转录组测序的结果,建立了 启动子序列-转录强度数据库,作为后续模型训练的数据集。
文献数据收集
枯草芽孢杆菌作为重要的原核模式微生物,近年来,研究者们也通过天然启动子之间的串联及融合,或者是对天然启动子的核心元件进行突变等策略构建了一系列枯草芽孢杆菌的人工启动子,用于枯草芽孢杆菌基因的表达调控。
通过查阅文献,我们全面搜集了此前已在枯草芽孢杆菌中构建合成启动子文库的相关数据[1-9]。相关文献中的启动子强度多以实验中的绿色荧光蛋白表达强度的相关强度表征,但由于文献中数据信息类型多样且存在缺少,需要进一步的补充与处理。根据我们收集到的数据的不同类型,分别采取以下策略进行完善:
- 类型一:文章中有短序列信息且注明起始位点:
处理方式:利用blast检索短序列,补充上游序列至长度为61bp; - 类型二:文章中没有相对活性值,但提供柱状图:
处理方式:利用Getdata软件取点读近似值,获得启动子强度信息; - 类型三:文章中有启动子序列,但未标明转录起始位点:
处理方式:利用Softberry工具预测转录启动子转录起始位点,序列不足61bp 的利用Blast检索补足。
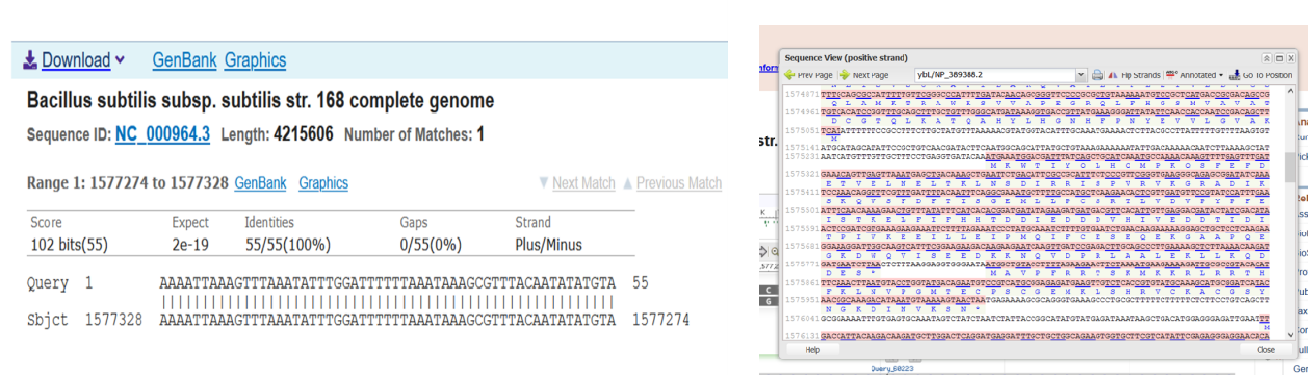
图5 利用blast检索补充启动子序列(类型一)
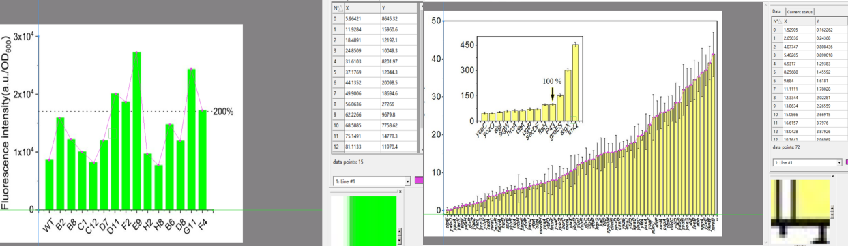
图6 利用Getdata软件读取启动子强度信息(类型二)
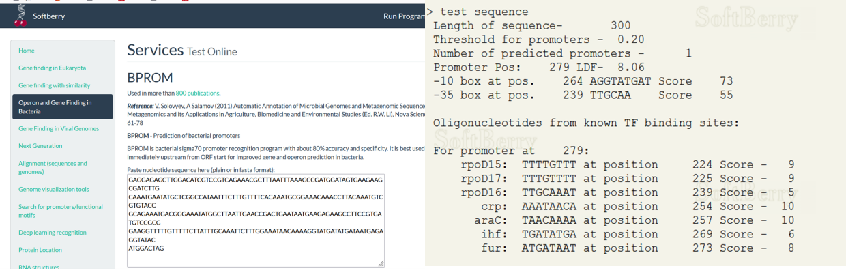
图7 利用Softberry工具预测转录起始位点(类型三)
文献启动子表征
经过上述的文献调研与整理,我们搜集得到了9篇文献12组共约600条启动子的序列与其荧光表达强度的数据,且均是以测量枯草芽孢杆菌中GFP荧光表达强度为表征手段。但由于不同实验室在培养微生物和测量荧光强度的实验设备、操作流程上存在差异,所以不同文献中的荧光值并不能直接使用。Jason等[10]研究发现,虽然荧光表达强度不能直接通用,但各启动子的相对表达强度是较稳定的。基于这一点,我们从每组数据中各选取一条启动子,以及枯草芽孢杆菌中常用启动子PvegI作为内标,共13条启动子在本地进行表征,从而获得13条启动子的相对强度关系,并根据每条启动子在其来源文库的数据将相对强度关系扩展,从而获得所有启动子的相对强度关系,构建序列-荧光强度数据库,具体实验流程如下:
一、含GFP空白质粒的构建
以Pad-123质粒为模板,以Pad-F和Pad-R为引物反向扩增得到线性化载体Pad-123。以Pbad-GFP质粒为模板,以G-F和G-R为引物扩增得到携带核糖体结合位点(RBS)序列的GFP片段,核糖体结合位点序列为“AAGAAGGAGATATACAT”。将PCR扩增得到的GFP片段与线性化载体Pad-123进行连接,获得重组质粒Pad-123-GFP。

图8 空白质粒Pad-123-GFP结构示意图(类型三)
二、各启动子报告质粒的构建
均以Pad-123-gfp质粒为模板,用经设计的不同引物进行PCR。各引物上设计有对应的启动子序列,且上引和下引含有25bp的同源区,以供再次连接成环形质粒。
- 经PCR和酶消化后,得到含有不同启动子与GFP的线性化载体片段,之后进行消化、胶收。
- 胶回收经消化的产物后,将其采用化学转化法转化进入感受态大肠杆菌DH5α,进行同源重组,以将线性化载体重新连接为环形质粒,同时利用大肠杆菌进行质粒的扩增。
- 转化的感受态大肠杆菌涂布于LB固体平板上,筛选白色菌落,转接提取质粒,条带显示为阳性克隆送测序验证,即可得13个含不同启动子的报告质粒。

图9 各报告质粒构建过程示意图(扩增得到线性片段)(类型三)

图10 各报告质粒构建过程示意图(线性片段同源重组)
实验中选用的启动子编号及其序列如下:
| 1 | GTGTTTAAAATTAAAGTTTAAGATGTTGGATTTTTTAAATAAAGCGTTTACAATATATGTA |
| 2 | GTGTTTAAAATTAAAGTTTAAATATTTGGATTTTTTAAATAGCTGGGTTACAATATATGTA |
| 3 | GCATGAAACTTTTCACCCATTTTTCGGTGATAAAAACATTTTCTTTTGATAAACTGAACGG |
| 4 | GTGTTTAAAATTAAAGTTTAAAGCGGGTGATTTTTTAAATAAAGCGTTTACAATATATGTA |
| 5 | AACGTTGATACCGGTTAAATTTTATTTGACAAAAATGGGCTCGTGTTGTCTAATAAATGTG |
| 6 | GCCTGCAAACGTTAAGATGGCAAGCTTGACAAGTATTTCCGACACATTTAGAATGAAGTTG |
| 7 | TTTATCCTTTACTGCGTCAATACACGTTGACACTCTTTTGATTTACTGTTAAATTATCAGG |
| 8 | GTGCTTAAAATTAAAGTTTAAATATTTGACATTTTTAAATAAAGCGTTTATAATATATGTA |
| 9 | ACGACATTTCATATTTCTTCTAATGCAGAAGATATGATAGAATAGAACTCGAACCGTATAT |
| 10 | GTTGGGCAGCCATCCTTGTTGGATTACTATGTATTTCGGCTCAATATTATTATGGTTAAAG |
| 11 | TTATCCTAACAGATCCATTGTTCATTGTAAATAATGGAATACGAGTTTTATATTATAAAAG |
| 12 | AAGGCAGGCAAATGCGAAAAAGGTGTTGACAACAGTGAATGCTTATGGTATAATTAGTGAA |
| Pvegi | TTTACCGAAACTTGCGGAACATAATTGAGGAATCATAGAATTTTGTCAAAATAATTTTATT GACAACGTCTTATTAACGTTGATATAATTTAAATTTTATTTGACAAAAATGGGCTCGTGTT GTACAATAAATGTAGTGAGGTGGATGCA |
三、报告菌株的构建
将各报告质粒及空白质粒(表征时作为参比)采用spizizen转化法转化进入B. subtilis168菌株,转化的枯草芽孢杆菌涂布于LB固体平板上,筛选白色菌落,条带显示为阳性克隆,即得13个含不同启动子的报告菌株。
Spizizen转化法详细流程如下:
- 使用接种环挑一个单菌落到不带相应抗性的5 ml GM1培养基的试管中,220 r/min、37 ℃过夜培养。
- 使用移液枪吸取500 μL菌液加到GM1培养基中(保证总体积5 mL不变,后同),220 r/min、37 ℃培养至对数生长期中后期(4.5 h)。
- 使用移液枪吸取1.5 mL菌液加入至现配好的GM2培养基中,220 r/min、37 ℃培养90 min。
- 使用移液枪吸取200 μL菌液置于无菌EP管中,加入适量质粒,置于37℃恒温培养箱培养1 h。
- 将EP管置于220 r/min、37 ℃培养1 h。
- 将菌液涂布于带有相应抗性的筛选平板中,37 ℃静置培养。
四、荧光表达强度的表征
将构建好的报告菌株进行发酵培养,在合适的时间测量其OD600和荧光强度,并以Pvegi为内标,以不含启动子的空白质粒为参比,进行一致化处理,详细流程如下
- 挑取平板活化的单克隆接入5 mL LB培养基中,37 ℃、220 r/min条件下过夜培养。
- 取适量培养物转接入含25 mL LB培养基的150 mL摇瓶中,使得初始OD600为0.5,在同样的条件下培养。每个样品3个平行。
- 首先测定细胞的OD600(4-6),4000rpm离心2 min,弃上清,等量双蒸馏水重悬细胞,重复离心重悬一次,彻底洗去培养基。
- 将200 μL样品转移到酶标板上,在485 nm激发后,在533 nm处测量gfp荧光。 关于所得数据的处理与结果,我们将在结果中作详细讨论,您可 点击此处进行浏览。
参考文献
[1]Xu J, Liu X, Yu X, et al. Identification and characterization of
sequence signatures in the Bacillus subtilis promoter P ylb for tuning promoter strength[J].
Biotechnology letters, 2020, 42(1): 115-124.
[2]Han L, Cui W, Suo F, et al. Development of a novel strategy for robust synthetic bacterial
promoters based on a stepwise evolution targeting the spacer region of the core promoter in
Bacillus subtilis[J].
Microbial cell factories, 2019, 18(1): 1-14.
[3]Song Y, Nikoloff J M, Fu G, et al. Promoter screening from Bacillus subtilis in
various conditions hunting for synthetic biology and industrial applications[J]. PloS one, 2016,
11(7): e0158447.
[4]Yu X, Xu J, Liu X, et al. Identification of a highly efficient stationary phase promoter in
Bacillus subtilis[J]. Scientific reports, 2015, 5(1): 1-9.
[5]Guiziou S, Sauveplane V, Chang H J, et al. A part toolbox to tune genetic expression in
Bacillus subtilis[J]. Nucleic acids research, 2016, 44(15): 7495-7508.
[6]Liu X, Wang H, Wang B, et al. High-level extracellular protein expression in Bacillus
subtilis by optimizing strong promoters based on the transcriptome of Bacillus subtilis
and Bacillus megaterium[J].
Protein expression and purification, 2018, 151: 72-77.
[7]Zhou C, Ye B, Cheng S, et al. Promoter engineering enables overproduction of foreign proteins
from a single copy expression cassette in Bacillus subtilis[J]. Microbial cell factories,
2019, 18(1):
1-11.
[8]Yang S, Du G, Chen J, et al. Characterization and application of endogenous phase-dependent
promoters in Bacillus subtilis[J]. Applied microbiology and biotechnology, 2017, 101(10):
4151-4161.
[9]Yuan P, Sun G, Cui S, et al. Engineering a ComA Quorum-Sensing circuit to dynamically control
the production of Menaquinone-4 in Bacillus subtilis[J]. Enzyme and Microbial Technology,
2021, 147: 109782.
[10]Coelho R V, Dall'alba G, De Avila E Silva S, et al. Toward Algorithms for Automation of
Postgenomic Data Analyses: Bacillus subtilis Promoter Prediction with Artificial Neural
Network [J]. Omics-a
Journal of Integrative Biology, 2020, 24(5): 300-9.